Questions? 800-523-5874 | [email protected]
- Prepmaster™ Specimen Preparation Robot
- TEM Grids
- TEM Window Grids
- Omniprobe Nanomanipulation Systems
- K-kit Wet "Liquid" TEM Kit
- Specimen Mounts
- SEM Specimen Holders
- Index and Finder SEM Grids
- SEM for Forensics
- SEM Sample Preparation Station Materials
- Cryogenic Personal Protection Equipment
- Cryo Dewars & Flasks
- Cryogenic Grids & Accessories
- Cryogenic Vials & Racks
- Cooling Chambers & Ice Baths
- Prepmaster™ Specimen Preparation Robot
- Laboratory Microwave Ovens
- LYNX II Automated Tissue Processor
- EMS Poly III
- Microtomes
- Tissue Slicers
- Rapid Immersion Freezer
- Heaters & Chillers
- SEM Cooling Stage
- Glow Discharge Systems
- Sputter Coaters & Carbon Coaters
- Stages
- Freeze Dryers
- Critical Point Dryers
- Cryo-SEM Preparation System
- Specimen Transfer Systems
- Decontaminators
- Desiccators
- Centrifuges
- Dry Baths
- Stirrers, Hot Plates
- Vortexers & Magnetic Mixers
- Rotators & Rockers
- Ovens & Incubators
- Vibration Isolation
- Air Sampling
- Vacuum Pumps
Filters
- Ice
- Overnight Shipping Only
- Shelf Life
Conventional ImmunoGold Reagents
Conventional Immunogold Reagents are available in four size classes. The monodisperse size population makes the conjugates suited for multiple labeling with no overlap. The Conventional Immunogold Reagents are the classical conjugates in immuno electron microscopy; they are a good choice when the antigen is abundant and the accessibility of the antigen is relatively good.
Introduction
The conventional labeling approach in transmission and scanning electron microscopy utilizes secondary immunogold reagents based on particles that can be observed without enhancement. These conjugates are suited for single and multiple labeling in electron microscopy, when the number of antigens available for binding is such that a relevant signal can be obtained.
The AURION Conventional Immunogold Reagents are built around colloidal gold particles with sizes of 6, 10, 15 or 25 nm. The particle population is monodisperse and thus shows minimal size variation and overlap. Typically, the coefficient of variance for the 6 and 25 nm particle size conjugates is less than 12%, whereas the 10 and 15 nm size conjugates show less then 10% variation.
The table below lists a few physical characteristics of gold conjugates.
| Particle Diameter | #Au atoms | MWt.(daltons) | #particles/ ml | #Ab/(part.) |
| 6 nm | 6500/td> | 1.3·106 | 2.4·1013 | 1-2 |
| 10 nm | 30·103 | 6·106 | 5·1012 | 7-12 |
| 15 nm | 100·103 | 20·106 | 1.5·1012 | 25-40 |
| 25 nm | 470·103 | 92·106 | 3.3·1011 | 115-180 |
Features of Conventional Immuno Gold Reagents
- for single and multiple labeling
- gold particle sizes of 6, 10 ,15 and 25 nm
- monodisperse particle population
- minimal size variation and overlap
- OD520 nm of 1.0 to warrant cluster free storage
- coefficient of variance: <12% for the 6 and 25 nm conjugates and <10% for the 10 and 15 nm conjugates
Product Description
AURION Conventional Immunogold Reagents are tailored to contain 10-20 µg of specific protein/ml. The reagents are supplied in PBS with 1% Bovine Serum Albumin and 15 mM NaN3 at an OD520nm of 1.0 to warrant prolonged cluster free storage. The activity of each lot is determined using a dot-spot test system as described by Moeremans et al., J. Immunol. Methods, 74, (1984), 353. Actual lot specifications (size, variation and expiry date) are indicated on the accompanying package insert.
Regular package: for the labeling of 1000-2000 grids
Small package: for the labeling of 400-800 grids
Specificity
Aurion offers the widest range of Conventional Immunogold Reagents.
AURION Conventional Immunogold Reagents are prepared using the highest quality antibodies or binding agents available. All antibodies are immuno affinity purified and immuno cross-adsorbed to reduce non-specific reactions.
Storage
AURION Conventional Immunogold Reagents have a guaranteed shelf life of 18 months from the date of quality control analysis.
The products should be stored at 4-8°C. Freezing is not recommended.
Technical Tips
Conventional ImmunoGold Reagents Application Instructions
Reactivity of Protein A and Protein
Overview Micrographs FAQs Workshops Custom Labeling Protocols Newsletters

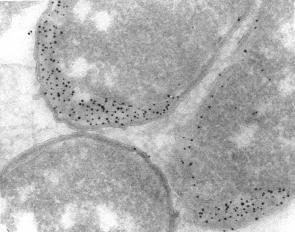 Immunolabeling of the periplasmic space in ultrathin cryosections of Escherichia coli with a protein A gold conjugate. Courtesy M. de Jong
Immunolabeling of the periplasmic space in ultrathin cryosections of Escherichia coli with a protein A gold conjugate. Courtesy M. de Jong 2nm")
Goat-anti-Rabbit IgG (H&L) 2nm
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25105 | Description: Goat anti Rabbit IgG 2nm | MSDS | Pack: 1 mL | Price: $429.50 |
Add to Quote:

|
|
|
| Storage:4-8°C | Cat #: 25106 | Description: Goat anti Rabbit IgG 2nm | MSDS | Pack: 0.4 mL | Price: $246.00 |
Add to Quote:
|
|

Goat-anti-Rabbit IgG H&L
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25103 | Description: EM Goat/Rabbit IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25108 | Description: EM Goat/Rabbit IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25112 | Description: EM Goat/Rabbit IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25115 | Description: EM Goat/Rabbit IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25104 | Description: EM Goat/Rabbit IgG, 6 nm | MSDS | Pack: 1 mL | Price: $246.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25109 | Description: EM Goat/Rabbit IgG, 10 nm | MSDS | Pack: 1 mL | Price: $246.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25113 | Description: EM Goat/Rabbit IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $246.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25116 | Description: EM Goat/Rabbit IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $246.00 |
Add to Quote:
|
|
")
Goat-anti-Mouse IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25123 | Description: EM Goat/Mouse IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25128 | Description: EM Goat/Mouse IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25132 | Description: EM Goat/Mouse IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25135 | Description: EM Goat/Mouse IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $429.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25124 | Description: EM Goat/Mouse IgG, 6 nm | MSDS | Pack: 1 mL | Price: $246.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25129 | Description: EM Goat/Mouse IgG, 10 nm | MSDS | Pack: 1 mL | Price: $246.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25133 | Description: EM Goat/Mouse IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $246.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25136 | Description: EM Goat/Mouse IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $246.00 |
Add to Quote:
|
|
")
Goat-anti-Mouse IgM (µ-chain)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25143 | Description: EM Goat/Mouse IgM, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25148 | Description: EM Goat/Mouse IgM, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25152 | Description: EM Goat/Mouse IgM, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25155 | Description: EM Goat/Mouse IgM, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25144 | Description: EM Goat/Mouse IgM, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25149 | Description: EM Goat/Mouse IgM, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25153 | Description: EM Goat/Mouse IgM, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25156 | Description: EM Goat/Mouse IgM, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|

Goat-anti-Mouse IgG+IgM
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25163 | Description: EM Goat/Mouse IgG/IgM, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25168 | Description: EM Goat/Mouse IgG/IgM, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25172 | Description: EM Goat/Mouse IgG/IgM, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25175 | Description: EM Goat/Mouse IgG/IgM, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25164 | Description: EM Goat/Mouse IgG/IgM, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25169 | Description: EM Goat/Mouse IgG/IgM, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25173 | Description: EM Goat/Mouse IgG/IgM, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25176 | Description: EM Goat/Mouse IgG/IgM, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
")
Goat-anti-Rat IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25183 | Description: EM Goat/Rat IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25188 | Description: EM Goat/Rat IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25192 | Description: EM Goat/Rat IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25195 | Description: EM Goat/Rat IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25184 | Description: EM Goat/Rat IgG, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25189 | Description: EM Goat/Rat IgG, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25193 | Description: EM Goat/Rat IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25196 | Description: EM Goat/Rat IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
")
Goat-anti-Human IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25203 | Description: EM Goat/Human IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25208 | Description: EM Goat/Human IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25212 | Description: EM Goat/Human IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25215 | Description: EM Goat/Human IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25204 | Description: EM Goat/Human IgG, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25209 | Description: EM Goat/Human IgG, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25213 | Description: EM Goat/Human IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25216 | Description: EM Goat/Human IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
")
Rabbit-anti-Goat IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25223 | Description: EM Rabbit/Goat IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25228 | Description: EM Rabbit/Goat IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25232 | Description: EM Rabbit/Goat IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25235 | Description: EM Rabbit/Goat IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25224 | Description: EM Rabbit/Goat IgG, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25229 | Description: EM Rabbit/Goat IgG, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25233 | Description: EM Rabbit/Goat IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25236 | Description: EM Rabbit/Goat IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|

Goat-anti-Biotin
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25243 | Description: EM Goat/Biotin, 6 nm | MSDS | Pack: 2.5 mL | Price: $468.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25248 | Description: EM Goat/Biotin, 10 nm | MSDS | Pack: 2.5 mL | Price: $468.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25252 | Description: EM Goat/Biotin, 15 nm | MSDS | Pack: 3.5 mL | Price: $468.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25255 | Description: EM Goat/Biotin, 25 nm | MSDS | Pack: 3.5 mL | Price: $468.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25244 | Description: EM Goat/Biotin, 6 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25249 | Description: EM Goat/Biotin, 10 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25253 | Description: EM Goat/Biotin, 15 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25256 | Description: EM Goat/Biotin, 25 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|

| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25263 | Description: EM Streptavidine/Gold, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25268 | Description: EM Streptavidine/Gold, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25272 | Description: EM Streptavidine/Gold, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25275 | Description: EM Streptavidine/Gold, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25264 | Description: EM Streptavidine/Gold, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25269 | Description: EM Streptavidine/Gold, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25273 | Description: EM Streptavidine/Gold, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25276 | Description: EM Streptavidine/Gold, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|

Protein A
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25282 | Description: EM Protein-A/Gold, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25284 | Description: EM Protein-A/Gold, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25286 | Description: EM Protein-A/Gold, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25288 | Description: EM Protein-A/Gold, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25283 | Description: EM Protein-A/Gold, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25285 | Description: EM Protein-A/Gold, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25287 | Description: EM Protein-A/Gold, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25289 | Description: EM Protein-A/Gold, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|

Biotinylated Albumin
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25298 | Description: EM Biotinylated Albumin/Gold, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage: | Cat #: 25299 | Description: EM Biotinylated Albumin/Gold, 10 nm | MSDS | Pack: 1.0 mL | Price: $261.00 |
Add to Quote:
|
|

Mouse Monoclonal anti-FITC Gold Conjugates
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25582 | Description: Mouse-Anti-FITC,10nm | MSDS | Pack: 2.5 mL | Price: $503.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25583 | Description: Mouse-Anti-FITC,10nm | MSDS | Pack: 1 mL | Price: $288.50 |
Add to Quote:
|
|

Goat-anti-Chicken IgG
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25586 | Description: Goat-Anti-Chicken IgG, 6nm | MSDS | Pack: 2.5 mL | Price: $503.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25588 | Description: Goat-Anti-Chicken IgG, 10nm | MSDS | Pack: 2.5 mL | Price: $503.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25590 | Description: Goat-Anti-Chicken IgG, 15nm | MSDS | Pack: 3.5 mL | Price: $503.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25592 | Description: Goat-Anti-Chicken IgG, 25nm | MSDS | Pack: 3.5 mL | Price: $503.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25587 | Description: Goat-Anti-Chicken IgG, 6nm | MSDS | Pack: 1 mL | Price: $288.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25589 | Description: Goat-Anti-Chicken IgG, 10nm | MSDS | Pack: 1 mL | Price: $288.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25591 | Description: Goat-Anti-Chicken IgG, 15nm | MSDS | Pack: 1.4 mL | Price: $288.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25593 | Description: Goat-Anti-Chicken IgG, 25nm | MSDS | Pack: 1.4 mL | Price: $288.50 |
Add to Quote:
|
|

Protein G
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25312 | Description: EM Protein-G/Gold, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25314 | Description: EM Protein-G/Gold, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25316 | Description: EM Protein-G/Gold, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25318 | Description: EM Protein-G/Gold, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25313 | Description: EM Protein-G/Gold, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25315 | Description: EM Protein-G/Gold, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25317 | Description: EM Protein-G/Gold, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25319 | Description: EM Protein-G/Gold, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
")
Goat-anti-Guinea Pig IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25323 | Description: EM Goat/Guinea Pig IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25328 | Description: EM Goat/Guinea Pig IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25332 | Description: EM Goat/Guinea Pig IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25335 | Description: EM Goat/Guinea Pig IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $445.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25324 | Description: EM Goat/Guinea Pig IgG, 6 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25329 | Description: EM Goat/Guinea Pig IgG, 10 nm | MSDS | Pack: 1 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25333 | Description: EM Goat/Guinea Pig IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25336 | Description: EM Goat/Guinea Pig IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $261.00 |
Add to Quote:
|
|
")
Rabbit-anti-Sheep IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25343 | Description: EM Rabbit/Sheep IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25348 | Description: EM Rabbit/Sheep IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25352 | Description: EM Rabbit/Sheep IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25355 | Description: EM Rabbit/Sheep IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25344 | Description: EM Rabbit/Sheep IgG, 6 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25349 | Description: EM Rabbit/Sheep IgG, 10 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25353 | Description: EM Rabbit/Sheep IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25356 | Description: EM Rabbit/Sheep IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
 2 Fragment of Goat-anti-Rabbit IgG (H&L)")
F(ab') 2 Fragment of Goat-anti-Rabbit IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25363 | Description: EM F(Ab')2, Goat/Rabbit IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25362 | Description: EM F(Ab')2, Goat/Rabbit IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25366 | Description: EM F(Ab')2, Goat/Rabbit IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25368 | Description: EM F(Ab')2, Goat/Rabbit IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25364 | Description: EM F(Ab')2, Goat/Rabbit IgG, 6 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25365 | Description: EM F(Ab')2, Goat/Rabbit IgG, 10 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25367 | Description: EM F(Ab')2, Goat/Rabbit IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25369 | Description: EM F(Ab')2, Goat/Rabbit IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
 2 Fragment of Goat-anti-Mouse IgG (H&L)")
F(ab') 2 Fragment of Goat-anti-Mouse IgG (H&L)
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25373 | Description: EM F(Ab')2, Goat/Mouse IgG, 6 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25372 | Description: EM F(Ab')2, Goat/Mouse IgG, 10 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25376 | Description: EM F(Ab')2, Goat/Mouse IgG, 15 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25378 | Description: EM F(Ab')2, Goat/Mouse IgG, 25 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25374 | Description: EM F(Ab')2, Goat/Mouse IgG, 6 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25375 | Description: EM F(Ab')2, Goat/Mouse IgG, 10 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25377 | Description: EM F(Ab')2, Goat/Mouse IgG, 15 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25379 | Description: EM F(Ab')2, Goat/Mouse IgG, 25 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
 2 Fragment of Goat-anti-Mouse IgG+IgM")
F(ab') 2 Fragment of Goat-anti-Mouse IgG+IgM
| Storage | Cat # | Description | MSDS | Pack | Price | Quote | Quantity | |
|---|---|---|---|---|---|---|---|---|
| Storage:4-8°C | Cat #: 25383 | Description: F(Ab')2, Goat/Mouse IgG/IgM 6 nm | MSDS | Pack: 2.5 mL | Price: $464.50 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25382 | Description: F(Ab')2, Goat/Mouse IgG/IgM 10 nm | MSDS | Pack: 2.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25386 | Description: F(Ab')2, Goat/Mouse IgG/IgM 15 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25388 | Description: F(Ab')2, Goat/Mouse IgG/IgM 25 nm | MSDS | Pack: 3.5 mL | Price: $496.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25384 | Description: F(Ab')2, Goat/Mouse IgG/IgM 6 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25385 | Description: F(Ab')2, Goat/Mouse IgG/IgM 10 nm | MSDS | Pack: 1 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25389 | Description: F(Ab')2, Goat/Mouse IgG/IgM 25 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
|
| Storage:4-8°C | Cat #: 25387 | Description: F(Ab')2, Goat/Mouse IgG/IgM 15 nm | MSDS | Pack: 1.4 mL | Price: $263.00 |
Add to Quote:
|
|
- 1
- 2